The DNA sequence data generated by high-throughput sequencing technology is short in length and the amount of data is very large. The challenges and opportunities of big data in high-throughput sequencing environment were analyzed. The research results of data compression, metagenomic data sequence splicing, and algorithm and tools for metagenomic data sequence analysis were summarized and discussed. Finally, the development trend of DNA short-read sequence data under high-throughput sequencing is prospected.
High throughput sequencing analysisHigh-throughput sequencing, parallel sequencing of millions to billions of DNA molecules at a time, also known as next-generation sequencing technology, enables in-depth, detailed, and comprehensive analysis of the transcriptome and genome of a species, so Also known as deep sequencing. Mainly include: High-throughput Sequencing, Next Generation Sequencing, Deep Sequencing.
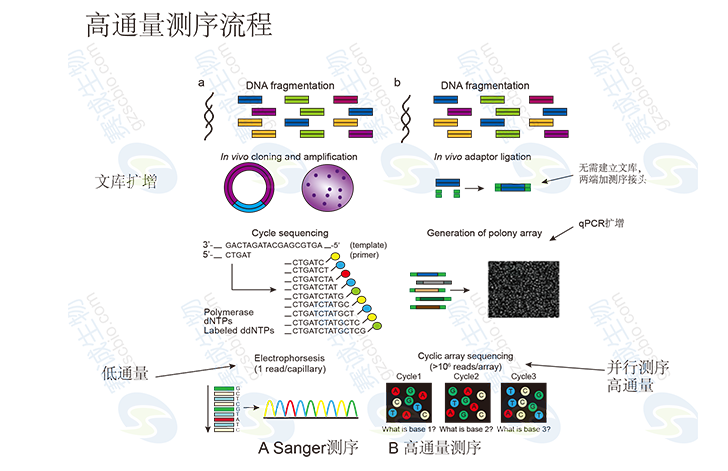
Figure 1 High-throughput sequencing process
High-throughput sequencing applications are available in a wide range of applications:1 DNA sequencing: genome-wide de novo sequencing, genome resequencing, metagenomic sequencing, human exome capture sequencing.
2 RNA sequencing: transcriptome sequencing, small RNA sequencing, electronic expression profiling.
3 Epigenome study: ChIP-Seq, DNA methylation sequencing.
Genomic sequencing
Genomic sequencing is the high-throughput sequencing of the genomic DNA of the species. According to whether there are known genomic data, it is mainly divided into de novo whole genome sequencing and genome resequencing. De novo genome sequencing is to de novo sequencing the species of unknown genomic sequence, using bioinformatics analysis to splicing and assembling the sequence to obtain the genomic map of the species. Whole genome resequencing is the sequencing of different individuals' genomes for species of known genomic sequences, and based on this, differential analysis of individuals or groups.
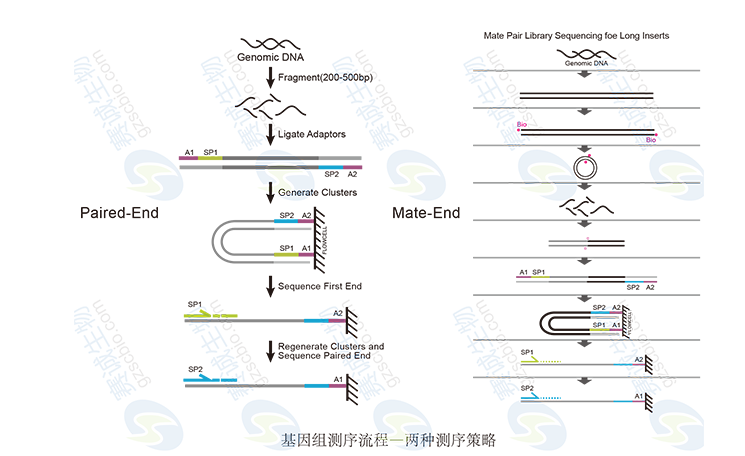
Figure 2 Genomic sequencing strategy
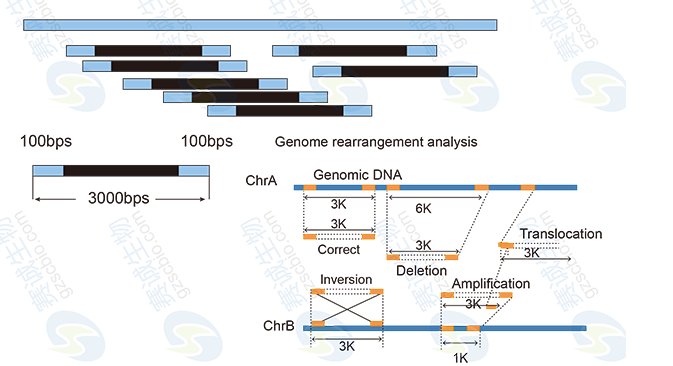
Figure 3 Paired-end principle
Paired-End method, after the genome is interrupted, a certain length (200-500 bp) sequence is selected to link the two ends of the linker for sequencing. Mate-end is more complex. After the sequence is interrupted, a certain length sequence (3-5kb) is selected. Biotin should be connected first, then cyclized, then interrupted, biotin enriched, and the two ends are ligated for sequencing at both ends. .
Genomic sequencing uses bioinformatics to analyze the results, mainly covering the following aspects.
1 Data output processing: image recognition and Base Calling\ remove joint sequences, detect and remove pollution sequences, etc.
2 genome assembly: raw data statistics, sequencing depth analysis, assembly results statistics, etc.;
3 genome annotations: Coding Gene annotations, RNA classification annotations, repeated sequence annotations, etc.;
4 Gene function annotation: GO function classification, Interpro function classification, etc.;
5 Comparative genomic and molecular evolution analysis: SNP/InDel/CNV detection.
Metagenome sequencingMetagenomic sequencing is the sequencing of all microorganisms in a particular environment, such as the intestines, soil, seawater, etc. This method can detect microbial species and dominant species in the environment, revealing microbial community diversity, population structure, evolutionary relationship, functional activity, mutual cooperation and relationship with the environment. Many microorganisms in the natural environment cannot be isolated and cultured, and this method does not require isolation and cultivation of microorganisms. The metagenomic sequencing method now has genome-wide metagenomic sequencing and 16S/18S rRNA metagenomic sequencing.
1 genome-wide metagenomic sequencing
High-throughput sequencing technology enables complete genome-wide metagenomic sequencing of total DNA from environmental samples, enabling species classification studies, community structure, phylogenetic evolution, functional annotation, and metabolic network studies between species. The value of genetic resources to develop new microbial active substances. Compared with the traditional Sanger method, the speed is fast, the cost performance is high, the cycle is short, and the sequencing amount of a single sample can be close to saturation.
The analysis of metagenomic sequencing information mainly includes: splicing assembly, species classification composition analysis, gene prediction and functional annotation, generating Profiling table, principal component analysis (PCA), screening factors significantly related to sample grouping, and comparing and analyzing multiple samples.
2 16S/18S rRNA macrogenomic sequencing
16S/18S rRNA is the most commonly used target molecule for microbial community analysis and bacterial evolution research and classification research. The next generation sequencing technology is used to sequence the variable region of 16S/18S rDNA without cloning and screening. Species composition of microbial populations, true species distribution and abundance information.
16S/18S rRNA sequencing information analysis mainly includes: species classification, species abundance analysis, OTU (Operational Taxonomic Units) analysis, diversity analysis, phylogenetic analysis, comparative analysis between multiple samples.
Human exome capture sequencingAn exome refers to a collection of all exon regions that contain important information needed to synthesize proteins, covering most of the functional variations associated with an individual's phenotype. Compared with whole genome resequencing, exome sequencing only needs to target the DNA of the exon region, with deeper coverage, higher data accuracy, and more simple, economical and efficient.
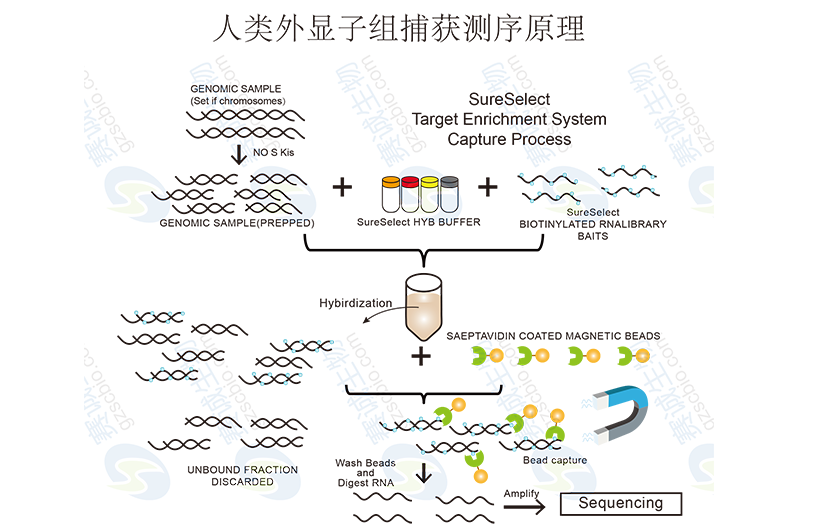
Figure 4 Principle of human exome capture sequencing
Exon capture refers to hybridization of exon chips, capture of the genomic exon sequence, and sequencing of the captured sequence. Commonly used exons are now Roche NimbleGen Sequence Capture 2.1M Human Exome Array and Agilent SureSelect Target Enrichment System (Human Exome).
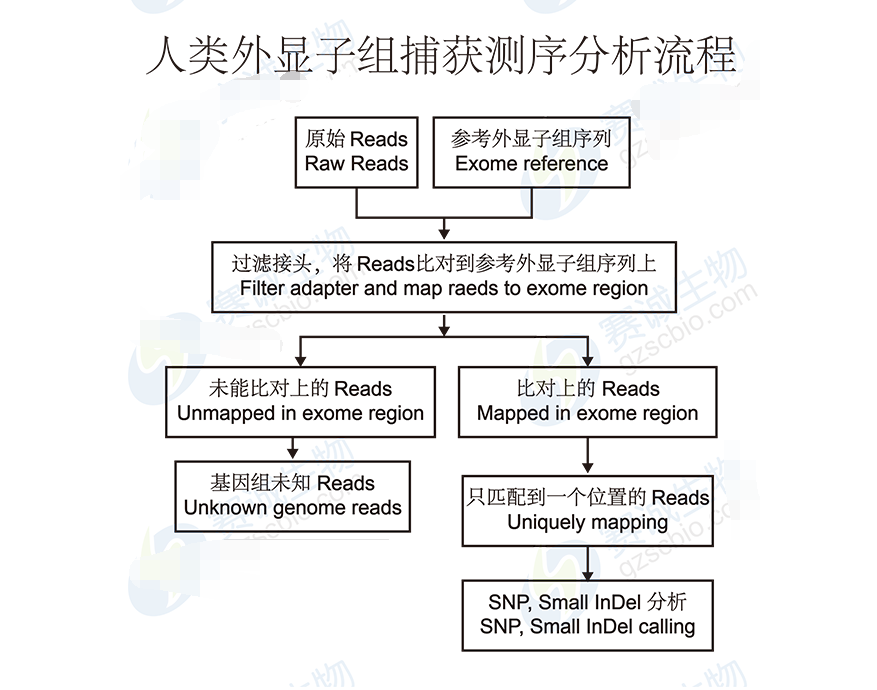
Figure 5 Human exome capture sequencing analysis process
Transcriptome sequencingA transcriptome is the sum of all RNAs that a particular cell can transcribe under a certain functional state, including mRNA and non-coding RNA.
The second-generation sequencing system accurately detects a single base and is not interfered with by prior information in the study. Researchers can quickly obtain almost all mRNA transcript sequences in a particular organ or tissue of a certain species, thereby enabling Development: UTRs region definition, variable shear studies, low-abundance new transcript findings, fusion gene identification, cSNP (coding sequence single nucleotide polymorphism) studies.
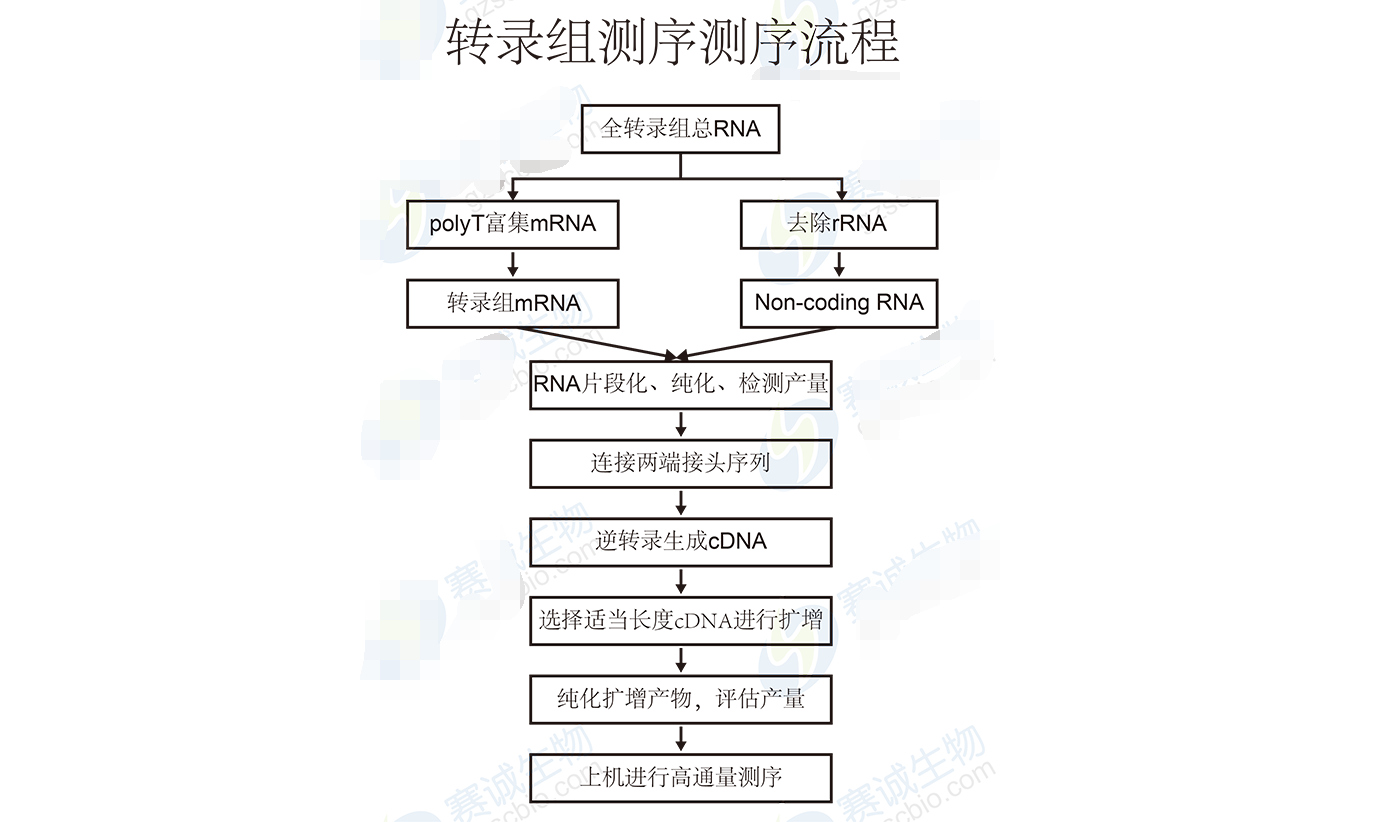
Figure 6 Transcriptome sequencing process
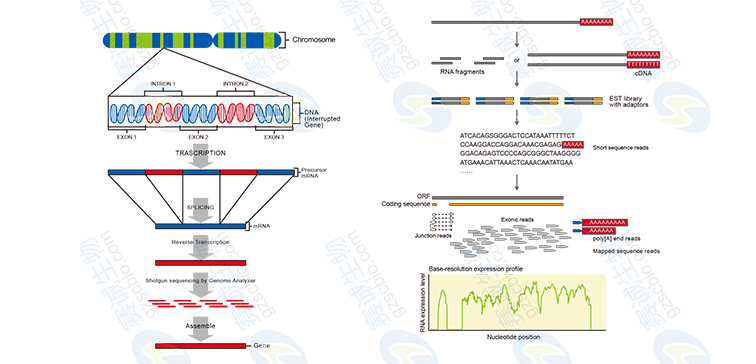
Figure 7: No reference sequence and reference sequence transcriptome sequencing process
Non-reference sequence transcriptome analysis includes: 1 sequencing data yield statistics, data components and quality assessment; 2 Contig and Scaffold length distribution; 3 Unigene length distribution and functional annotation, GO classification, Pathway analysis, differential expression analysis; 4 protein function Prediction and classification, differentially expressed genes, GO enrichment and Pathway enrichment analysis.
The transcriptome analysis with reference sequence includes: 1 basic data statistics, comparison of reference sequences; 2 sequence distribution on the genome; 3 sequencing depth analysis, randomness assessment and gene differential expression analysis; 4 new gene prediction, gene alternative splicing Identification and gene fusion identification.
Electron expression profilingDigital Gene Expression (DGE) is also known as mRNA tag profiling, also known as Tag-SAGE. The principle is to sequence a 21 nt sequence tag in the gene by two enzyme digestions. Because its sequencing only targets the expressed genes, the amount of data generated is relatively small, which is an economical and rapid research method for studying gene expression profiles. It is a genome-wide gene expression profile under specific processing conditions and has been widely used in research fields such as functional genomics and medicine.
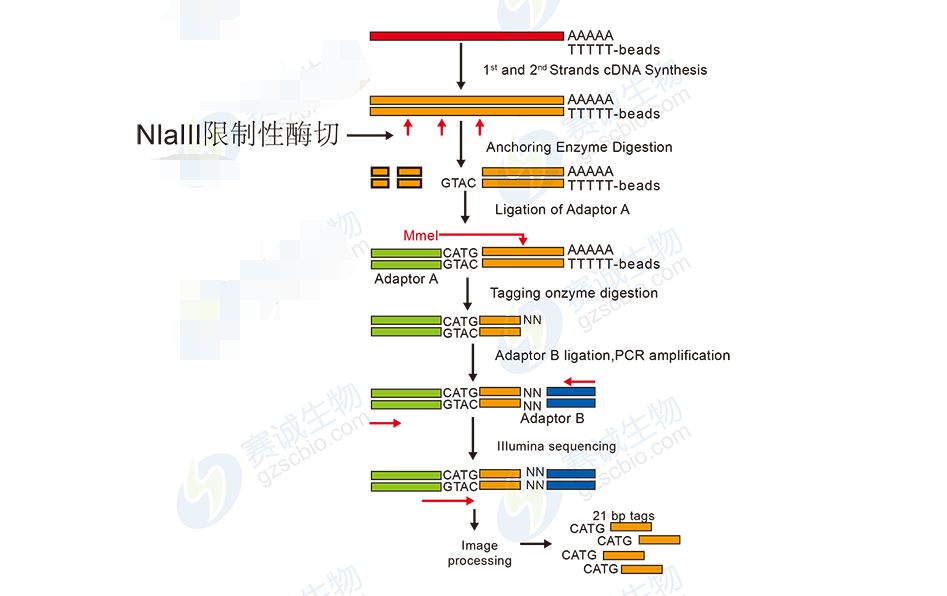
Figure 8 Electronic expression spectrum sequencing flow chart
The electronic expression profiling includes: image recognition and original base data reading, decontamination, de-joining, tag sequence counting statistics, genomic alignment and statistics, gene sequence alignment to obtain the expressed gene list, gene differential expression analysis, Clustering and expression type analysis, GO gene enrichment and classification analysis, Pathway enrichment and classification analysis, protein interaction network analysis, antisense strand transcript and new transcript detection.
Small RNA sequencingSmall RNA refers to endogenous non-protein-encoding RNA of 21-31 nt in length, which is widely present in higher and lower organisms, and regulates the life processes such as transcription and post-transcriptional levels of mRNA. Small RNAs are now known to be grouped into three categories: microRNAs (miRNAs), small interfering RNAs (siRNAs), and RNAs interacting with piwi (piRNAs).
The miRNA is 21 to 24 nt in length and is produced from a transcript of a typical stem-loop secondary structure (pri-miRNA), which plays an important role in the degradation and inhibition of target mRNA of plants and animals. siRNA, which is 19 to 25 nt in length, is produced by long double-stranded RNA and plays an important role in the degradation and inhibition of target mRNA of plants and animals. piRNA, 26~31 nt in length, is defined by the Piwi protein that interacts with it, and current studies indicate that it plays a role in the formation of gametes.
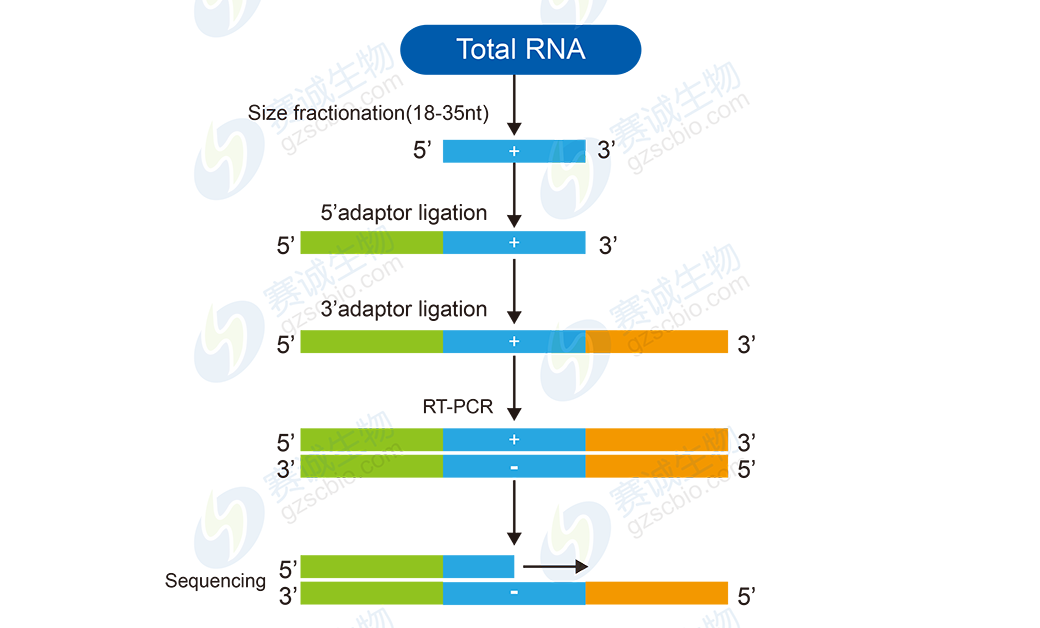
Figure 9 Small RNA sequencing flowchart
Small RNA sequencing analysis includes the following two main aspects:
1 Basic analysis: raw data reading, de-joining, decontamination sequences, length distribution statistics, genomic alignment, etc.
2 Advanced analysis: classification of Small RNA, identification of miRNA / siRNA / piRNA, prediction of new miRNAs, cluster analysis of differentially expressed miRNAs, etc.
ChIP-Seq
ChIP-Chromatin Immunoprecipitation refers to the precipitation of proteins that interact with chromatin, such as histones, transcription factors, etc., by antibodies, and the acquired DNA sequences. ChIP-Seq is the sequencing of the sequences obtained by ChIP by high-throughput sequencing for protein-DNA interaction studies.
The ChIP-Seq analysis includes:1 ChIP Sequencing results were compared to reference genome sequences.
2 ChIP Sequencing reads in the genome-wide distribution: the unique alignment of the distribution of reads in the regions of the repeats, the only comparison of the distribution of reads on the functional elements of each gene, the only comparison of the full genome coverage depth of the reads.
3 genome-wide peak scan: peak scan, peak length distribution statistics, full genome coverage of peak, distribution of peak on gene functional elements,
4 Peak-related gene analysis screening and GO function enrichment analysis.
Difference analysis of more than 5 samples: Difference analysis based on peak-related genes, peak-based difference analysis.
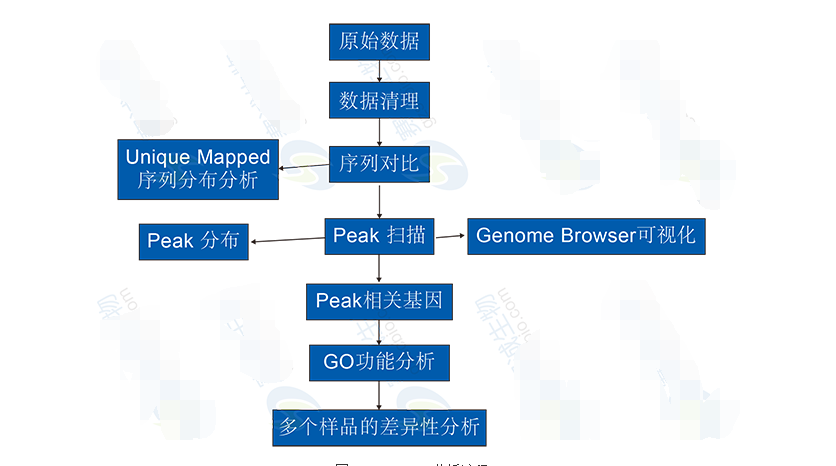
Figure 10 ChIP-Seq analysis process
DNA methylation sequencingDNA methylation plays an important role in the regulation of body development and gene expression, and has a great correlation with the occurrence and development of various cancers. Therefore, research on genomic DNA methylation has been a hot topic. There are two main methods for studying DNA methylation by high-throughput sequencing. One is MeDIP, which is co-immunoprecipitated by an antibody that binds to a DNA methylation site, and then the resulting DNA sequence is sequenced. The other is Bisulfite Sequencing, which treats the genome by Bisulfite to distinguish methylation sites.
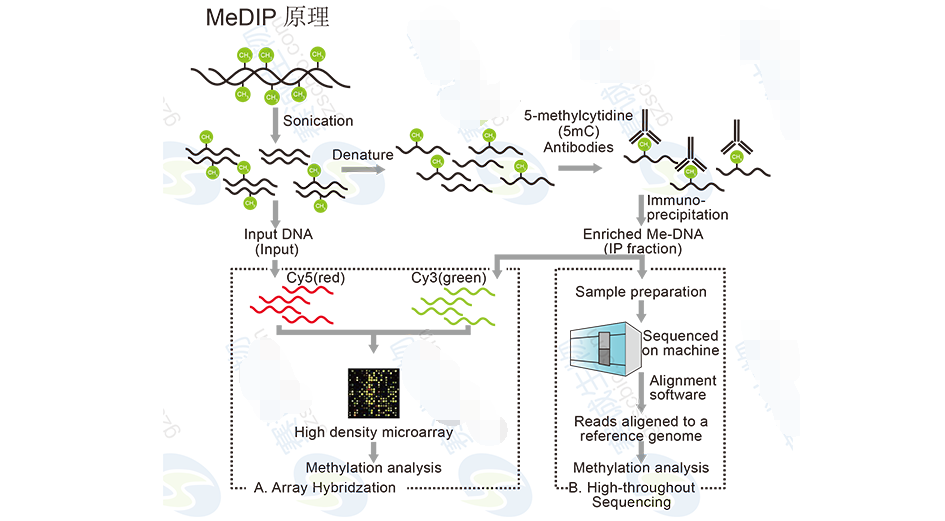
Figure 11 MeDIP principle
The MeDIP-Seq analysis includes:1 Comparison of the MeDIP-seq sequence with the reference sequence.
2 MeDIP-seq sequence data distribution trends across the genome: MeDIP-seq sequencing reads distribution on each chromosome across the genome, MeDIP-seq sequencing reads coverage depth across the genome, MeDIP-Seq sequencing reads in CG, Coverage depth at CHG and CHH sites, distribution of MeDIP-Seq sequencing reads on different gene functional elements, and distribution of MeDIP-Seq sequencing reads in different OE content regions.
3 Statistics of MeDIP-seq sequence enrichment region (peak): Peak scan, Peak length quantity and proportional distribution statistics, OE content distribution statistics of individual sample Peak, search for Peak related genes, and statistics of Peak distribution on different gene functional elements .
4 Peak-based multi-sample difference analysis: Analyze the Peak-related differential gene between the two samples, and perform GO functional enrichment analysis and pathway function analysis on the differential genes between the two samples.
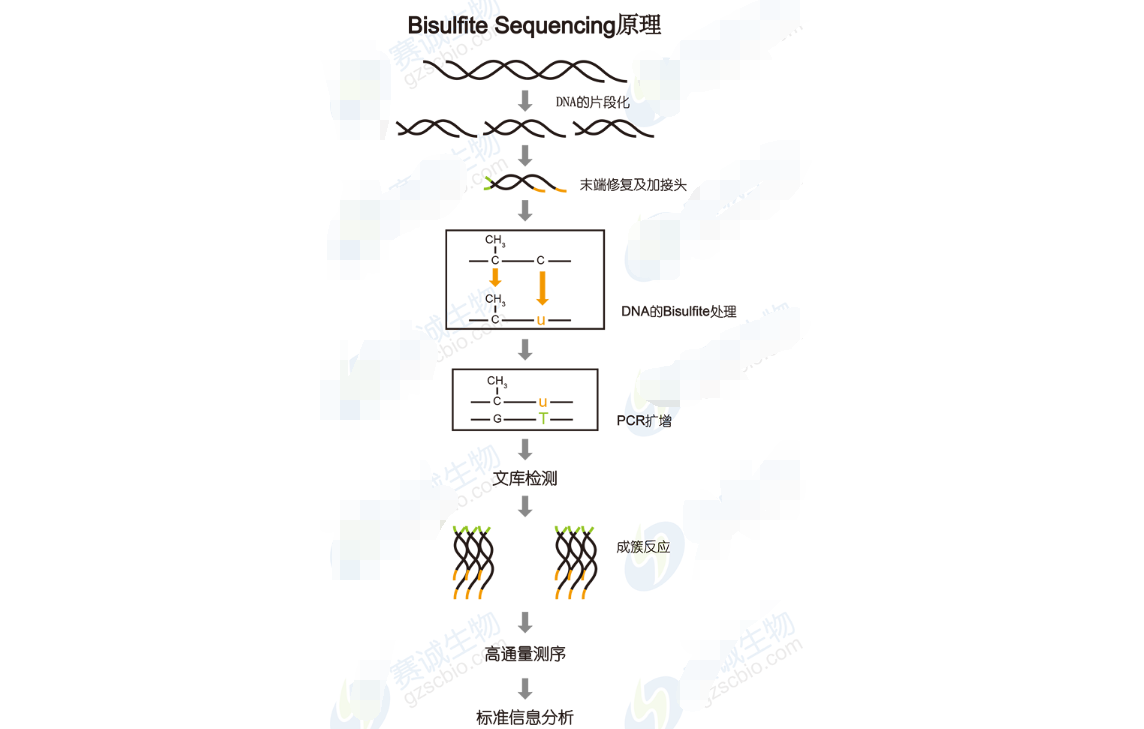
Figure 12 Bisulfite Sequencing principle
Bisulfite Sequencing analysis includes:1 Alignment of the Bisulfite-seq sequence with a reference sequence.
2 Depth and coverage analysis: cumulative distribution of C-base effective sequencing depth, genome coverage at different reads sequencing depth.
3 Calculate the methylation level of the C base.
4 Trend analysis of genome-wide methylation data distribution: distribution ratio of CG, CHG and CHH in methylated C bases (H=A, C or T), methylation level of all C in CG, CHG and CHH The methylation level of C in CG, CHG and CHH in each chromosome (this analysis is currently only used for "human"), and the methylation level of C in CG, CHG and CHH in different gene regions, different genes The methylation level of C in CG, CHG and CHH in the element region, and the sequence characteristics of the 9 bp sequence near methylation C in CHG and CHH.
5 Whole genome DNA methylation map: density distribution of methylated C bases at the chromosome level (this analysis is currently only used for "human"), Scaffold methylation C base density distribution (this analysis is for species : non-human), methylation distribution characteristics of different genomic regions, DNA methylation levels in different transcription elements of the genome.
6 Differential methylation region (DMR) analysis.
·Basic precautions
Do not put expensive oil into low-quality cartridges to avoid wastage. Most pre-filled oil cartridges have the so-called 510 thread. The oil cartridge screws onto a rechargeable battery. Some of these batteries have buttons and some heat up automatically when you pump the oil. Some batteries have multiple temperature settings and some heat up to a preset temperature; these features need to be known in advance.
·Cleaning notes
Use a suitable cleaning tool to clean them, such as activated charcoal or dried tea leaves in a used pipe to absorb the oil. It is important not to use alcohol or other boiling water to clean the pipe, and to wait until it has cooled down completely before cleaning. Otherwise, the hot stem will come into contact with the watery liquid and cause the mouthpiece tenon to loosen, thus shortening the life of the 510 cartridges.
This is what you should be aware of when using 510 cartridges. At the same time, when using 510 cartridges, there are still some vaping tips, for example, when using them, be careful not to suck too hard, will not produce smoke. When you inhale too hard, the smoke is sucked directly into your mouth and not atomized by the atomizer, so gently inhaling is more powerful and gives you a better vaping experience.
510 Cartridge Oem,Leakproof 510 Cartridge,510 Battery And Cartridges Oem,510 Cartridge
Shenzhen MASON VAP Technology Co., Ltd. , https://www.cbdvapefactory.com